





MUTYH-associated polyposis (MAP) is an autosomal recessive hereditary cancer syndrome. It’s most commonly seen in people of northern European ancestry, where an estimated 1 in 20,000–40,000 have MAP (two pathogenic variants on opposite chromosomes) and 1%–2% have one MUTYH pathogenic variant.
Most disease is caused by two founder pathogenic variants, p.Tyr179Cys (p.Y179C) and p.Gly396Asp (p.G396D): together they account for 50%–82% of MUTYH variants identified in European patients with MAP. The two founder variants are available on some direct-to-consumer genetic tests (DTCGTs), and patients may bring positive findings to their healthcare providers. The interpretation and risks of pathogenic MUTYH variants require consideration of ethnicity, personal history of polyps and cancer, and family medical history as well as whether the findings are monoallelic (one altered copy) or biallelic (two altered copies on opposite chromosomes).
Cancer Risks and Clinical Presentation
MAP should be suspected when an individual has 20 or more colon polyps over a lifetime, which can include adenomas, hyperplastic, or sessile serrated polyps. Without surveillance, patients with biallelic pathogenic variants (homozygous) have increased risk for many cancers. Those with a monoallelic pathogenic variant (heterozygous) have an increased risk for colon cancer and possibly an increased risk for gastric, hepatobiliary, endometrial, and breast cancer. Screening recommendations are based on personal and family history as well as homozygous or heterozygous status.
Cancer Risk in People With Homozygous MAP
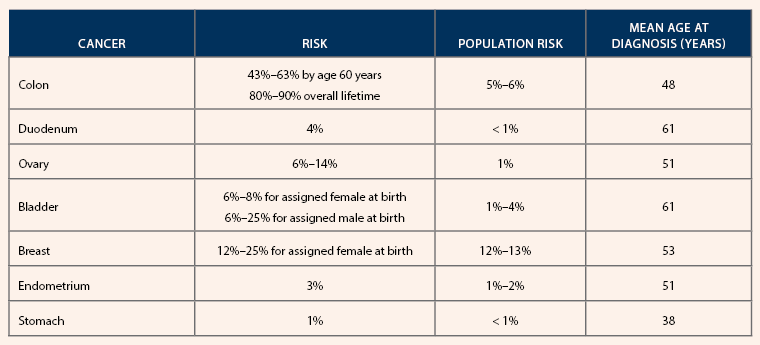
Note. Based on information from National Comprehensive Cancer Network and Nielsen et al.
Future Generations
Germline testing is typically offered at age 25 to coincide with screening initiation. Siblings of an individual with MAP have a 25% risk of having MAP and a 50% carrier risk. Other first-degree relatives (e.g., parents, children) also have a 50% carrier risk. Both biologic sexes are equally likely to inherit and pass on a pathogenic variant.
Nursing Implications
Nurses should review colonoscopy histories, assess patients’ lifetime cumulative polyps, and refer those with suspicious histories to a genetics professional for evaluation. DTCGT-identified pathogenic variants should be confirmed in a Clinical Laboratory Improvement Amendments–approved laboratory.
Reassure patients that the syndrome is manageable and provide accurate information and support, including rationale for prevention and detection guidelines. Because information about risks and best management is continually evolving, reevaluate patients’ care every 12–18 months to ensure they are adhering to the latest recommendations.
Screening Recommendations for Individuals With a MUTYH Pathogenic Variant
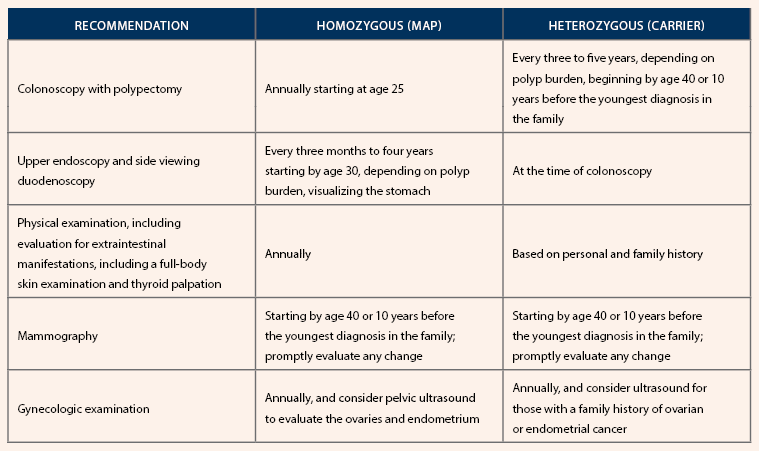
Note. Based on information from National Comprehensive Cancer Network, Nielsen et al., and Win et al.