





Since the first biosimilar agent was approved by the U.S. Food and Drug Administration (FDA) in 2015, patients and providers have had concerns about the implications for their care and practice, respectively. Because 6 of the 12 biosimilar drugs currently approved in the United States have indications for oncology practice, oncology nurses have a responsibility to understand the drugs’ safety and efficacy for the patients in their care.
To help oncology nurses stay updated on the latest information related to cancer biosimilars, the Clinical Journal of Oncology Nursing published a dedicated supplement to its October 2018 issue that explored the agents in depth. Tariman’s introductory article to the supplement reported on a literature review that found that clinical safety, efficacy, and tolerability were the top concerns for patients, prescribers, and clinicians as the new drugs make their way into practice.
What Are Biosimilar Drugs?
“All biosimilars are biologics, which are medicines derived from living systems that may be sourced from nature or produced using recombinant techniques,” Tariman wrote. They are also referred to as genetically engineered or biotech products. The drugs are bioequivalent to the cancer biologic agents already used in practice, such as monoclonal antibodies, but can be produced at a much lower cost, making them more affordable for patients who may be already facing financial toxicity related to their treatment.
Global biologic patent protection ended in 2006, and the United States and Europe began a new era of biosimilar development. Europe’s work has been mostly in therapeutic applications, such as vaccines and antitoxins, and those products came to market first. The United States led innovations in biotechnology and biologic therapies, particularly monoclonal antibodies for cancer and rheumatologic diseases, with its first biosimilar reaching the market in 2015.
As of October 2018, six biosimilar drugs are FDA approved for cancer-related indications:
- Bevacizumab-awwb
- Epoetin alfa-epbx
- Filgrastim-sndz
- Filgrastim-aafi
- Trastuzumab-dkst
- Pegfilgrastim-jmdb
Creating Biosimilars
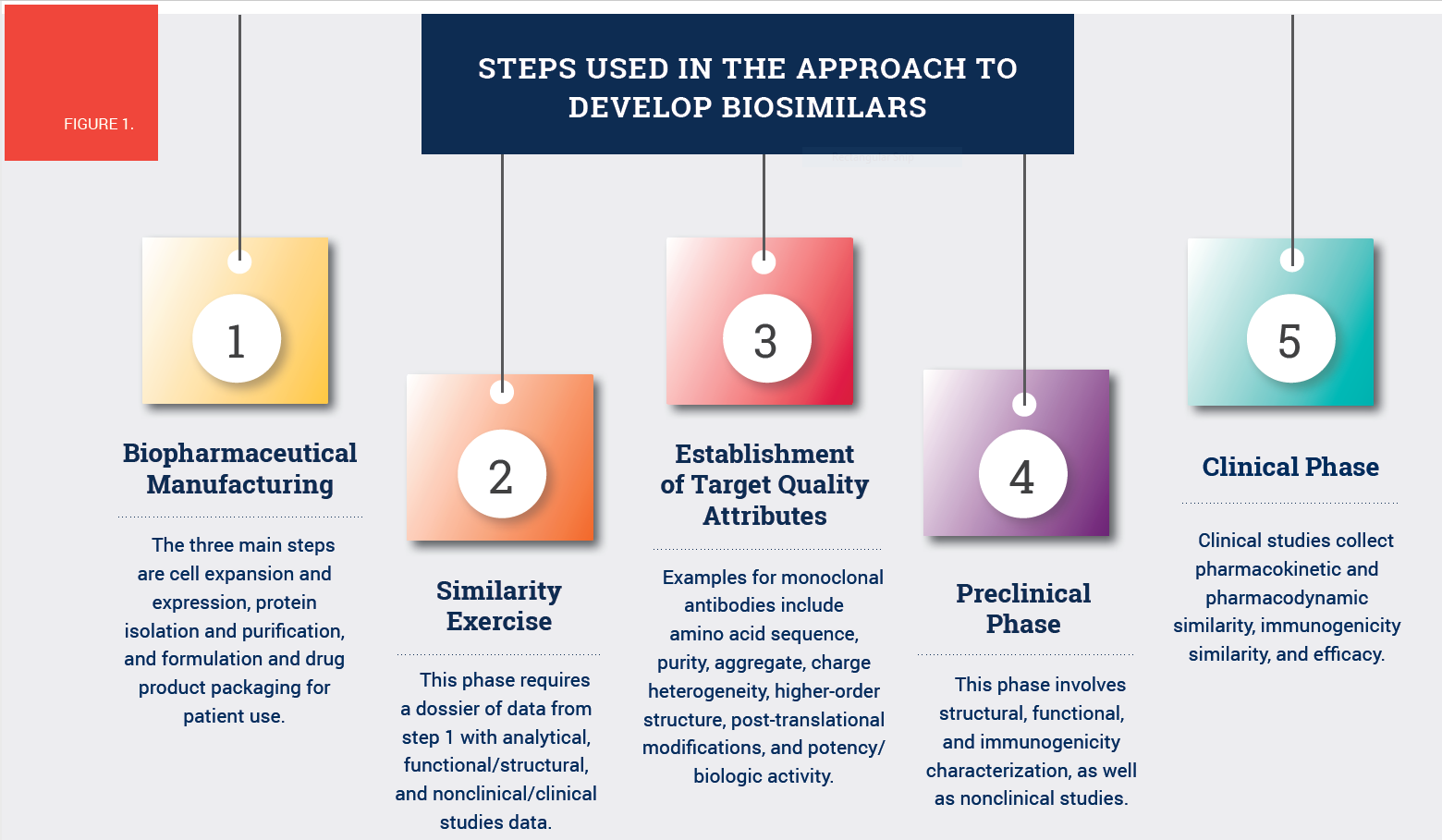
Both biosimilars and their biologic reference products are “complex molecules produced by biotechnology in living systems,” Tariman explained. The manufacturing process is intricate and highly controlled, and the products are assessed on preclinical, structural, and functional levels for clinical safety, efficacy, and immunogenicity. See Figure 1 for the approach to develop biosimilars.
Unlike synthetic generic drugs, biosimilars and their biologic reference products are larger (100–1,000 times the size) and more complex. Biosimilars are created commercially from genetically modified cell lines (e.g., bacteria, yeasts, murine hybridomas) to produce biologically equivalent copies of proteins. They are not identical to the reference product but rather reverse engineered based on the primary sequence of the reference biologic product in the public domain.
Biosimilars are unique from their reference product because of modifications to the manufacturing process (e.g., different choice of host cells) that may affect the protein structure, function, clinical pharmacology, immunogenicity, purity, and stability of the final product. For that reason, the drugs undergo state-of-the-art analytical techniques and quality assurance to ensure that they have no clinically meaningful differences from the reference products.
Testing and Quality Assurance
FDA and its European equivalent both use high similarity and comparability as the two standards for biosimilars. Analysis methods include peptide mapping (amino acid sequence attribute), cell-based assays (binding attribute), and in vivo assays (biologic activity attribute). Any differences found must be identified, analyzed, and reduced through repetitive processes and preclinical testing to protect patient safety and clinical efficacy.
To ensure immunogenicity, FDA’s approval pathway measures both binding and neutralizing antidrug antibodies because they are associated with lower trough concentration, reduced efficacy, and increased frequency of infusion reactions. The process to ensure bioequivalence is tailored to each individual reference biologic, although Tariman explained that tolerability and efficacy are the two highest priorities.
Implications for Practice
Because biosimilars can be created more efficiently than reference products, they are more affordable for un- or underinsured patients. More biosimilar agents may come to market as additional patents expire. Through interprofessional collaboration, oncology nurses can work with pharmacists, social workers, and physicians to deliver safe and effective care. Tariman emphasized the importance of pharmacovigilance and accurate reporting of safety issues and side effects.
For more information on biosimilars, refer to the Clinical Journal of Oncology Nursing’s biosimilars supplement.
This monthly feature offers readers a concise recap of full-length articles published in the Clinical Journal of Oncology Nursing (CJON) or Oncology Nursing Forum. This edition summarizes “Biosimilars: Exploring the History, Science, and Progress,” by Joseph D. Tariman, PhD, RN, ANP-BC, FAAN, the introductory article to a special supplement on biosimilars to the October 2018 issue of CJON. Questions regarding the information presented in this article should be directed to the CJON editor at CJONEditor@ons.org. Photocopying of this article for educational purposes and group discussion is permitted.